
Мкц лекарство инструкция: Целлюлоза микрокристаллическая МКЦ «Анкир®-Б» — инструкция по применению, аналоги, форма выпуска
Мкц таблетки 0,5 г: инструкция, цена, аналоги | таблетки Фармаком
Дополнительный источник пищевых волокон. Способствует снижению массы тела, улучшению липидного и углеводного обмена, выведению токсических веществ из организма. Имеет дезинтоксикационные свойства.
СОСТАВ
МКЦ, кальция стеарат.
ОПИСАНИЕ ДЕЙСТВУЮЩИХ ВЕЩЕСТВ
Микрокристаллическая целлюлоза (МКЦ) — производится из натурального растительного сырья и предназначена для очищения организма. Это ценный пищевой элемент, при помощи которого можно существенно уменьшить калорийность собственного рациона, при этом не теряя все полезные свойства еды. Пищевые волокна МКЦ не растворяются в воде и не расщепляются в желудке человека. Главное их свойство — способность связывать и выводить из организма тяжелые металлы, токсины, продукты распада, свободные радикалы.
Пищевые волокна играют важную роль в нормализации работы желудочно-кишечного тракта — они впитывают жидкость, разбухают, в несколько раз увеличиваясь в объеме, и, раздражая механорецепторы желудка, вызывают ощущение сытости и избавляют от чувства голода, тем самым предотвращая переедание — один из факторов ожирения. В тонком кишечнике очищают его слизистую оболочку, что ведет к улучшению пристеночного пищеварения и всасывающей функции кишечника. Раздражая рецепторы кишечника, усиливают его перистальтику, за счет чего ликвидируется застой пищевого комка. Пищевые волокна МКЦ также положительно воздействуют на микрофлору кишечника, создавая благоприятную среду для жизнедеятельности полезных бактерий, которые синтезируют необходимые для организма витамины группы В, витамин РР и витамин К. Пищевые волокна очищают организм: выводят продукты незавершенного пищеварения, сокращают проявление аллергических реакций, улучшают деятельность кишечника, улучшают обмен веществ, способствуют снижению избыточной массы тела. МКЦ имеет массу преимуществ. При регулярном применении способствует существенному снижению массы тела; качественно нормализует уровень сахара в крови; выводит токсины и холестерин; существенно повышает физическую выносливость; является источником пищевых волокон; нормализует работу органов пищеварения.
В тонком кишечнике очищают его слизистую оболочку, что ведет к улучшению пристеночного пищеварения и всасывающей функции кишечника. Раздражая рецепторы кишечника, усиливают его перистальтику, за счет чего ликвидируется застой пищевого комка. Пищевые волокна МКЦ также положительно воздействуют на микрофлору кишечника, создавая благоприятную среду для жизнедеятельности полезных бактерий, которые синтезируют необходимые для организма витамины группы В, витамин РР и витамин К. Пищевые волокна очищают организм: выводят продукты незавершенного пищеварения, сокращают проявление аллергических реакций, улучшают деятельность кишечника, улучшают обмен веществ, способствуют снижению избыточной массы тела. МКЦ имеет массу преимуществ. При регулярном применении способствует существенному снижению массы тела; качественно нормализует уровень сахара в крови; выводит токсины и холестерин; существенно повышает физическую выносливость; является источником пищевых волокон; нормализует работу органов пищеварения.
СПОСОБ ПРИМЕНЕНИЯ
Взрослым принимать по 3 таблетки 3 раза в сутки в течение месяца, за 20 минут перед едой, запивая водой, соком или кефиром.
Важна регулярность приема МКЦ и достаточное количество потребляемой жидкости ― не менее 1,5 л в сутки, включая жидкость в блюдах, напитках и др. После 10-дневного перерыва курс можно повторить. Курс приема — 30 дней.
ПРОТИВОПОКАЗАНИЯ
Индивидуальная чувствительность к компонентам, беременность, период лактации, дети до 12 лет, при обострении заболеваний желудочно-кишечного тракта.
ЭНЕРГЕТИЧЕСКАЯ ЦЕННОСТЬ ККАЛ/100 Г
12,8 ккал.
ПИЩЕВАЯ (ПИТАТЕЛЬНАЯ) ЦЕННОСТЬ В 100 Г ПРОДУКТА
Белки — 0,2 г; углеводы — 3 г; жиры — 0 г.
ФОРМА ВЫПУСКА
Таблетки по 500 мг № 100.
СРОК ГОДНОСТИ
36 месяцев.
МКЦ Анкир-Б Таблетки 500мг №100
Аптека Здравсити (Живика)
круглосуточно
Данные от: 05. 04.2023
04.2023
Кострома, ул. Советская, 1198(4942)32-67-46
180.00 р.
В наличии 2 шт.
эвалар
Аптека Здравсити (Живика)
круглосуточно
Данные от: 05.04.2023
Кострома, ул. Советская, 1198(4942)32-67-46
193.00 р.
В наличии 1 шт.
эвалар
Будь здоров!
с 09:00 до 19:00 (закрыто)
Данные от: 05.04.2023
Кострома, ул. Калиновская, 428(4942)45-24-08
200.00 р.
В наличии 1 шт.
эвалар
Аптека 100М
с 09:00 до 21:00 (закрыто)
Данные от: 06.04.2023
Кострома, ул. Поселковая, 378(4942)48-00-40
Поселковая, 378(4942)48-00-40
214.70 р.
193.23
10% пн, вт, ср с 9 до 12 по карте Родина
В наличии 1 шт.
эвалар
Аптека 100М
с 09:00 до 21:00 (закрыто)
Данные от: 06.04.2023
Кострома, ул. Поселковая, 378(4942)48-00-40
215.00 р.
193.50
10% пн, вт, ср с 9 до 12 по карте Родина
В наличии 1 шт.
эвалар
Аптека 100Мс 09:00 до 21:00 (закрыто)
Данные от: 06.04.2023
Кострома, ул. Поселковая, 378(4942)48-00-40
220.00 р.
198.00
10% пн, вт, ср с 9 до 12 по карте Родина
В наличии 1 шт.
эвалар
Аптека ‘Здоровье’
с 08:00 до 19:00 (закрыто)
Данные от: 05. 04.2023
04.2023
Кострома, Петрковский бул., 38(4942)42-03-70
220.00 р.
В наличии 1 шт.
эвалар
Алоэ
с 09:00 до 21:00 (закрыто)
Данные от: 06.04.2023
Кострома, ул. Евгения Ермакова, 98(4942)49-64-54 доб. 5542
234.00 р.
В наличии 1 шт.
эвалар
Алоэ
с 09:00 до 21:00 (закрыто)
Данные от: 06.04.2023
Кострома, ул. Свердлова, 127а8(4942)49-64-54 доб. 5234
234.00 р.
В наличии 1 шт.
эвалар
Максавит
с 08:00 до 21:00 (закрыто)
Данные от: 05.04.2023
Кострома, ул.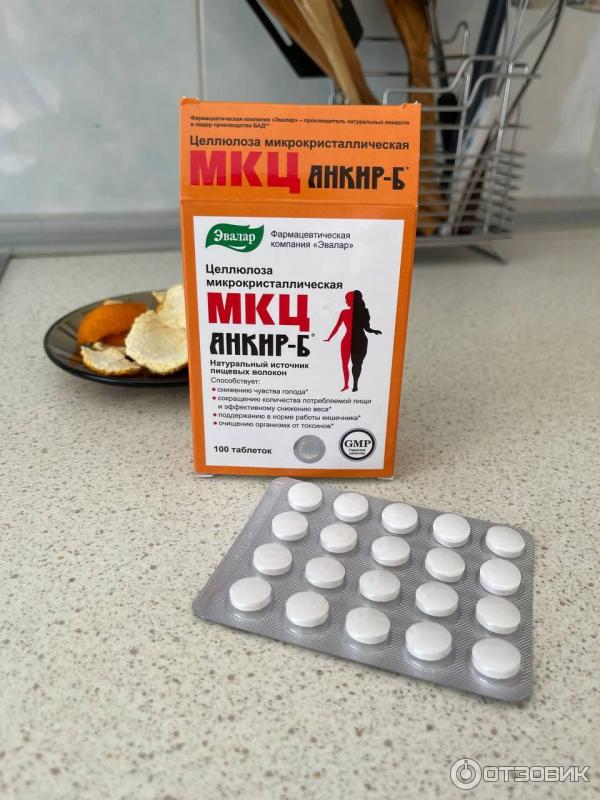 Советская, 798-920-398-08-71
Советская, 798-920-398-08-71
235.00 р.
В наличии 3 шт.
эвалар
Лидер-Фарм
с 09:00 до 22:00 (закрыто)
Данные от: 05.04.2023
Кострома, Давыдовский-3 мкрн, 11 (ТЦ Лента)8(4942)49-60-30
287.99 р.
В наличии 1 шт.
эвалар зао
Выводить по: 2050100Все
Как изучить и продать свое устройство
Как изучить и продать свое устройство
Медицинские устройства, продаваемые в США, подпадают под регулирующий контроль в соответствии с Федеральным законом о пищевых продуктах, лекарствах и косметических средствах (Закон FD&C) и положениями в Раздел 21- Свод федеральных правил (21 CFR), части 1-58, 800-1299. Регуляторный контроль и пути сбыта основаны на риске устройства. Регуляторный контроль необходим для обеспечения разумной гарантии безопасности и эффективности. Маркетинговые пути включают: Предпродажное уведомление (510(k)), Запрос на классификацию De Novo, Освобождение, Предпродажное одобрение (PMA), Протокол разработки продукта (PDP), Исключение для гуманитарного использования (HDE) и Заявка на получение лицензии на биопрепараты (BLA).
Регуляторный контроль необходим для обеспечения разумной гарантии безопасности и эффективности. Маркетинговые пути включают: Предпродажное уведомление (510(k)), Запрос на классификацию De Novo, Освобождение, Предпродажное одобрение (PMA), Протокол разработки продукта (PDP), Исключение для гуманитарного использования (HDE) и Заявка на получение лицензии на биопрепараты (BLA).
Предпродажные требования
Вывод устройства на рынок США может показаться сложным. Выполнение этих четырех шагов может помочь вам ориентироваться в этом процессе.
Четыре шага для вывода устройства на рынок:
- Шаг первый: Классифицируйте свое устройство и изучите применимые нормативные меры
- Шаг второй: Выберите и подготовьте правильное предпродажное представление
- Шаг третий: Отправьте предпродажную заявку в FDA и взаимодействуйте с персоналом FDA во время проверки
- Шаг четвертый:
Шаг первый: классифицируйте свое устройство и ознакомьтесь с применимыми мерами контроля
Первым шагом в подготовке устройства к продаже в США является определение того, как FDA классифицировало ваше устройство. Медицинское устройство определяется законом в разделе 201(h) Федерального закона о пищевых продуктах, лекарствах и косметике (FD&C).
Медицинское устройство определяется законом в разделе 201(h) Федерального закона о пищевых продуктах, лекарствах и косметике (FD&C).
Медицинские устройства относятся к одному из трех классов (I, II или III) в зависимости от степени риска, который они представляют. По мере того, как класс устройств увеличивается с класса I до класса II и класса III, регулирующий контроль также увеличивается, при этом устройства класса I подлежат наименьшему нормативному контролю, а устройства класса III подлежат наиболее строгому нормативному контролю. Классы устройств, нормативный контроль и типы подачи сведены в таблицу:
| Класс | Риск | Потенциальный вред | Регуляторный контроль | Тип представления или освобождение |
|---|---|---|---|---|
| Я | Самый низкий | Присутствует минимальный потенциал вреда | Общий | 510(к) 510(к) Освобожден |
| II | Умеренный | Более высокий риск, чем устройства класса I | Общие и специальные (при наличии) | 510(к) 510(к) Освобожден |
| III | Самый высокий | Поддерживают или поддерживают жизнь, имплантированы или представляют потенциальный необоснованный риск заболевания или травмы | Общий и PMA | ПМА |
Для получения дополнительной информации о нормативном контроле (общем и специальном), который может быть применим к вашему устройству, посетите веб-страницу нормативного контроля.
Следующие ресурсы могут помочь вам в определении классификации вашего устройства:
- База данных классификации продуктов FDA
- Панели классификации устройств
- Классифицируйте свое медицинское устройство
- Принадлежности для медицинских устройств
Если ваше медицинское устройство также является электронным продуктом, излучающим радиацию, оно также должно соответствовать применимым нормам 21 CFR, части 1000-1050.
Примечания:
- Если ваш продукт Комбинированный продукт — медицинское устройство плюс другой продукт, регулируемый FDA (например, лекарство, биологические препараты и т. д.), вам следует обратиться в Управление комбинированных продуктов (OCP) FDA по электронной почте по адресу: [email protected]. Основываясь на основном способе действия вашего продукта, OCP сообщит вам, в какой центр FDA вам нужно обратиться, чтобы продать ваш продукт.
- Даже если для вашего медицинского устройства не требуется предварительная отправка на рынок, вы все равно должны указать правильную классификацию вашего устройства, чтобы понять и соблюдать применимые нормативные требования.


Шаг второй: выберите и подготовьте правильное предпродажное представление
Вам следует выбрать и подготовить соответствующее предпродажное представление, если оно требуется для классификации вашего конкретного продукта. Для большинства медицинских устройств соответствующий тип представления указывается в классификации продуктов, которую можно получить из общедоступной базы данных классификации продуктов. Обратите внимание, что для некоторых типов устройств предварительная отправка не требуется. Для получения дополнительной информации см. веб-страницу исключений класса I/II. Если вы определили, что для вашего устройства не требуется предпродажная подготовка, вы можете пропустить второй и третий шаги и перейти к четвертому шагу. К наиболее распространенным типам предпродажной подачи относятся:
- 510(k) (предпродажное уведомление)
- PMA (предпродажное одобрение)
- Запрос на классификацию De Novo
- HDE (освобождение от гуманитарных устройств)
510(k)
Для некоторых устройств класса I и большинства устройств класса II требуется 510(k).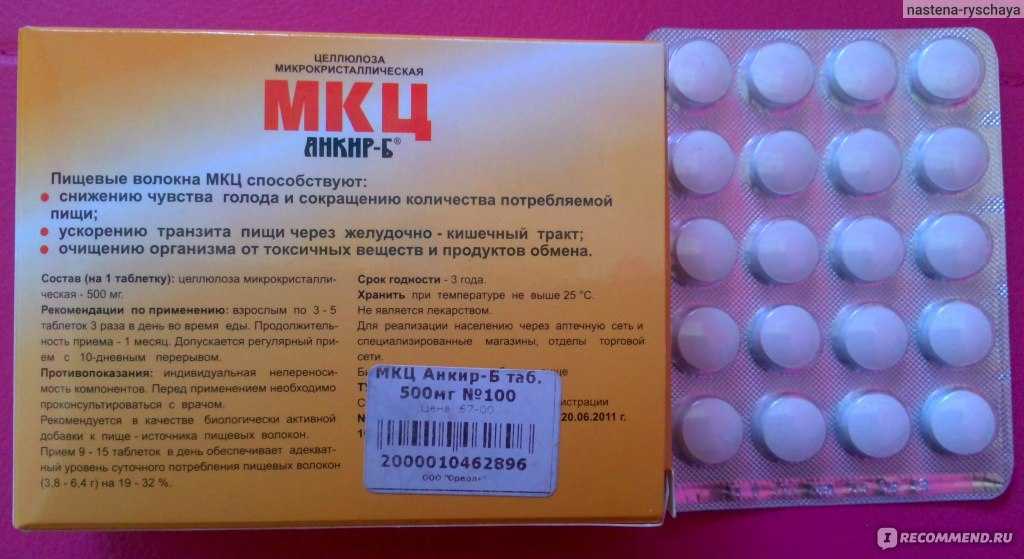 В 510(k) спонсор должен продемонстрировать, что новое устройство «по существу эквивалентно» предикатному устройству с точки зрения предполагаемого использования, технологических характеристик и тестирования производительности, если это необходимо. Информацию о том, как подготовить и отправить форму 510(k), см. в Предпродажном уведомлении Device Advice [510(k)].
В 510(k) спонсор должен продемонстрировать, что новое устройство «по существу эквивалентно» предикатному устройству с точки зрения предполагаемого использования, технологических характеристик и тестирования производительности, если это необходимо. Информацию о том, как подготовить и отправить форму 510(k), см. в Предпродажном уведомлении Device Advice [510(k)].
Некоторые устройства класса I и класса II освобождаются от требования об уведомлении 510(k), если они не превышают ограничения освобождения, указанные в 21 CFR xxx.9, где xxx относится к 21CFR 862-892. Например, эластичный бинт, классифицированный в соответствии с 21CFR 880.5075, освобождается от предпродажного уведомления при условии, что он не превышает ограничений, указанных в 21CFR 880.9.
PMA
Для устройств класса III требуется PMA. PMA является наиболее строгим типом предварительной подачи заявок. Прежде чем FDA одобрит PMA, спонсор должен предоставить действительные научные доказательства, демонстрирующие разумные гарантии безопасности и эффективности устройства по назначению.
Сведения о том, как подготовить и отправить PMA, см. в разделе Предпродажное одобрение рекомендаций по использованию устройств (PMA).
Запрос на классификацию De Novo
Процесс De Novo обеспечивает путь к классификации новых медицинских изделий, для которых только общие средства контроля или общие и специальные средства контроля обеспечивают достаточную уверенность в безопасности и эффективности для предполагаемого использования, но для которых нет предикатное устройство, продаваемое на законных основаниях. Для получения информации о том, как подготовить и подать запрос De Novo, обратитесь к следующим ресурсам:
- Закон FD&C, раздел 513(f)(2)
- Запрос на классификацию DeNovo (процесс De Novo)
- Оценка автоматического обозначения класса III (процесс De Novo) Резюме
HDE
HDE обеспечивает регуляторный путь для устройств класса III, предназначенных для пациентов с редкими заболеваниями или состояниями. Чтобы устройство имело право на HDE, спонсор должен сначала получить обозначение устройства для гуманитарного использования (HUD), которое предоставляется через заявку в Управление FDA по разработке продуктов для сирот (OOPD).
Информацию о том, как запросить разрешение на использование в гуманитарных целях, см. в разделе Назначение устройства для гуманитарного использования (HUD).
Для получения информации о том, как подготовить и подать заявку HDE, см. следующие ресурсы:
- 21CFR 814, подраздел H
- Руководство по программе освобождения от использования гуманитарных устройств (HDE)
Дополнительные ресурсы для помощи в подготовке предпродажной документации
FDA разработало несколько различных типов ресурсов, которые помогут вам подготовить предпродажную документацию. К ним относятся следующие:
- Отправка и отслеживание предпродажных заявок на медицинские устройства онлайн: зарегистрируйте учетную запись на портале CDRH для отправки предпродажных заявок CDRH eCopy или eSTAR онлайн
- CDRH Learn: серия обучающих модулей, семинаров и записанных веб-семинаров на основе видео, которые охватывают различные меры политики и руководства.
- CDRH’s Q-Submission Program: Потенциальные заявители будущих предрыночных заявок могут запросить отзыв в FDA через предварительную заявку, которая является частью программы Q-Submission. Мы рекомендуем вам ознакомиться с нашей онлайн-информацией и другими доступными ресурсами, прежде чем отправлять какие-либо запросы на обратную связь.
- CDRH Breakthrough Devices Program: Эта добровольная программа помогает пациентам получить более своевременный доступ к определенным медицинским устройствам, которые лечат или диагностируют опасные для жизни или необратимо изнурительные состояния, ускоряя их разработку, оценку и проверку. Устройства, на которые распространяются заявки на предварительное одобрение (PMA), предварительные уведомления [510(k)] или запросы на классификацию De Novo, могут иметь право на участие. Спонсоры могут в любое время отправить запрос на обозначение прорыва для своего устройства до отправки маркетинговой заявки на устройство.

Информация, которую следует учитывать при подготовке предпродажной документации
Средства контроля конструкции:
Устройства класса II и класса III должны разрабатываться в соответствии с средствами контроля конструкции в соответствии с Регламентом о системе качества (21 CFR 820. 30). Большинство устройств класса I не подпадают под действие Контроля за проектированием. Дополнительную информацию о средствах контроля конструкции см. в Руководстве по контролю конструкции для производителей медицинских устройств.
30). Большинство устройств класса I не подпадают под действие Контроля за проектированием. Дополнительную информацию о средствах контроля конструкции см. в Руководстве по контролю конструкции для производителей медицинских устройств.
Доклинические испытания: Типы информации и испытаний, необходимых для продажи вашего устройства, определяются многими факторами, включая классификацию устройства, принципы работы, технологические характеристики и маркировку. Доклинические испытания, проводимые в поддержку подачи заявки на допродажу медицинского изделия, должны соответствовать Надлежащим лабораторным практикам (GLP) в 21 CFR 58. Для получения дополнительной информации см. Рекомендуемое содержание и формат полных отчетов о тестировании для неклинических лабораторных испытаний производительности на допродаже. Материалы.
Стандарты консенсуса: FDA поощряет использование признанных FDA стандартов консенсуса при подаче заявок на продажу. Неотъемлемой частью наименее обременительного подхода к обзору устройств является опора на международные согласованные стандарты. Соответствие является добровольным, если только стандарт не включен посредством ссылки в нормативные акты (см. Документы Федерального реестра стандартов медицинских устройств). Руководство, Надлежащее использование стандартов добровольного консенсуса в предпродажных представлениях для медицинских устройств , объясняет, как использовать стандарты при отправке устройств, и описывает документацию, необходимую для поддержки их использования, включая декларации о соответствии и отчеты об испытаниях. По вопросам, касающимся надлежащего использования согласованных стандартов, вы можете обращаться по электронной почте по адресу [email protected].
Соответствие является добровольным, если только стандарт не включен посредством ссылки в нормативные акты (см. Документы Федерального реестра стандартов медицинских устройств). Руководство, Надлежащее использование стандартов добровольного консенсуса в предпродажных представлениях для медицинских устройств , объясняет, как использовать стандарты при отправке устройств, и описывает документацию, необходимую для поддержки их использования, включая декларации о соответствии и отчеты об испытаниях. По вопросам, касающимся надлежащего использования согласованных стандартов, вы можете обращаться по электронной почте по адресу [email protected].
FDA рекомендует производителям рассмотреть возможность участия в программе добровольной схемы аккредитации для оценки соответствия (ASCA). Эта добровольная пилотная программа предназначена для уменьшения нагрузки, связанной с отдельными предпродажными заявками, за счет повышения последовательности и предсказуемости в подходе FDA к оценке соответствия стандартам ASCA в предпродажных проверках медицинских устройств. FDA выпустило три окончательных руководящих документа, касающихся добровольной программы ASCA, которые можно загрузить с веб-страницы ASCA. По вопросам, касающимся добровольной программы ASCA, вы можете обращаться по адресу [email protected].
FDA выпустило три окончательных руководящих документа, касающихся добровольной программы ASCA, которые можно загрузить с веб-страницы ASCA. По вопросам, касающимся добровольной программы ASCA, вы можете обращаться по адресу [email protected].
Клинические доказательства: PMA, HDE и некоторые 510(k)s и запросы на классификацию De Novo требуют клинических доказательств. Перед началом клинического исследования спонсору исследования может потребоваться получить разрешение FDA на получение разрешения на использование исследовательского устройства (IDE). Исследование также должно быть одобрено соответствующим Институциональным наблюдательным советом (IRB). Клинические исследования должны соответствовать всем применимым правилам IDE и надлежащей клинической практике (GCP). Для получения дополнительной информации о правилах IDE и GCP см. Исключение для исследовательских устройств (IDE).
Маркировка: Маркировка устройства должна быть написана в соответствии с правилами маркировки и включена в предпродажную заявку.
Для получения информации о маркировке устройств см. следующие ресурсы:
- Маркировка 21 CFR 801
- Система маркировки устройств и уникальная система идентификации устройств (UDI)
Управление по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) создало уникальную систему идентификации устройств (UDI) для надлежащей идентификации медицинских устройств при их распространении и использовании. Правило UDI стало окончательным в сентябре 2013 года, и в течение нескольких лет оно будет вводиться поэтапно, главным образом на основе классификации устройств. После полного внедрения система UDI предложит ряд преимуществ для промышленности, FDA, потребителей, поставщиков медицинских услуг и систем здравоохранения, включая повышение безопасности пациентов и послепродажный надзор. Дополнительные сведения о требованиях к UDI см. в следующих источниках:
- Уникальный идентификатор устройства (UDI)
- Глобальная база данных UDI (GUDID)
- CDRH Learn (модули UDI)
Шаг третий: подготовьте соответствующую информацию для предпродажной подачи
После того, как вы подготовили соответствующую предпродажную заявку для вашего устройства, вам необходимо отправить ее в FDA и взаимодействовать с персоналом FDA во время ее рассмотрения. Прежде чем отправить заявку в FDA, вы должны знать следующее:
Прежде чем отправить заявку в FDA, вы должны знать следующее:
- Плата за использование медицинского устройства: Существует плата за использование, связанная с отправкой определенных маркетинговых приложений. Информацию о том, какие приложения подлежат оплате с пользователей, а также о сборах за использование этих приложений, см. в разделе Сборы с пользователей медицинских устройств.
- Программа определения малого бизнеса (SBD): Предприятие, которое квалифицировано и сертифицировано как «малое предприятие», имеет право на существенное снижение большинства этих сборов с пользователей. Дополнительную информацию о программе SBD см. в документе «Сниженная плата за пользование медицинскими устройствами: программа определения малого бизнеса (SBD)».
- Электронная копия: Для получения подробной информации о том, как подготовить электронную копию, см. Программу электронной копии для подачи заявок на медицинское оборудование.
- eSTAR: eSTAR — это интерактивная форма в формате PDF, которая помогает заявителям в процессе подготовки комплексной заявки на медицинское устройство. Для получения дополнительной информации см.: Добровольная программа eSTAR.

После того, как ваше заявление будет получено FDA, вы должны знать следующее:
- Административная проверка: После получения предпродажной заявки FDA проводит административную проверку, чтобы оценить, является ли заявка достаточно полной, чтобы ее можно было принять для рассмотрения по существу. Пожалуйста, ознакомьтесь с Политикой отказа в принятии для 510 (k) s, Проверками приема и подачи заявок на допродажное утверждение (PMA) или Проверка приемлемости для запросов на классификацию De Novo и программы освобождения от гуманитарных устройств (HDE): Руководство для промышленности и Управления по санитарному надзору за качеством пищевых продуктов и медикаментов. Персонал.
- Интерактивная проверка: Пока заявка находится на рассмотрении, сотрудники FDA общаются с заявителями, чтобы повысить эффективность процесса рассмотрения. См.: Типы коммуникаций во время рассмотрения заявок на медицинское оборудование.
- Отправка и отслеживание предпродажных заявок на медицинские устройства в Интернете: Портал CDRH. Когда вы отправляете заявку CDRH 510(k) (традиционную, специальную и сокращенную 510(k)s) на рассмотрение, ваш официальный корреспондент может отслеживать прогресс FDA в режиме онлайн в простой, лаконичный формат.
 См.: Типы коммуникаций во время рассмотрения заявок на медицинское оборудование.
См.: Типы коммуникаций во время рассмотрения заявок на медицинское оборудование.Шаг четвертый: Соблюдение применимых нормативных требований, включая регистрацию предприятий и список устройств
Регулятивные меры — это требования, основанные на оценке рисков, которые применяются к медицинским устройствам и обеспечивают надзор FDA для обеспечения разумной безопасности и эффективности медицинских устройств.
Устройства всех трех классов (класс I, II и III) подлежат общему контролю, если только они не освобождены от правил, которые частично требуют, чтобы предприятия по производству устройств: (1) регистрировали свои предприятия и составляли список медицинских устройств, которые они продают с FDA; (2) производить свои устройства в соответствии с Надлежащей производственной практикой; (3) маркировать свои устройства в соответствии с правилами маркировки; и (4) нельзя фальсифицировать или неправильно маркировать. Если устройство освобождено от одного из общих мер контроля, такое освобождение указано в правилах классификации для этого устройства.
Если устройство освобождено от одного из общих мер контроля, такое освобождение указано в правилах классификации для этого устройства.
Предприятие по производству устройств должно зарегистрировать свое предприятие и перечислить свои устройства в FDA. Информацию о том, как зарегистрироваться и составить список, можно найти на странице Регистрация и список устройств.
Если устройство требует предварительной подачи на рынок до выхода на рынок (т. е. медицинское устройство не является исключением), предприятия по производству устройств должны дождаться получения разрешения или одобрения FDA, прежде чем регистрировать и вносить в список.
Регистрация предприятия по производству устройств, присвоение регистрационного номера или внесение медицинского устройства в список никоим образом не означает одобрение или одобрение предприятия или его продукции FDA.
Получение торговой лицензии ЕС, шаг за шагом
Содержание
- Действия перед подачей заявки
- Подача заявки
- Оценка заявки
- Решение Европейской комиссии о регистрационном удостоверении
- Добровольное распределение намерений по выходу на рынок: пилотный проект
Европейское агентство по лекарственным средствам (EMA) отвечает за научную оценку заявок на получение централизованных регистрационных удостоверений в Европейском союзе (ЕС). Эта процедура авторизации позволяет фармацевтическим компаниям подавать в EMA единую заявку на получение разрешения на продажу и продавать лекарство и делать его доступным для пациентов и медицинских работников на всей территории Европейской экономической зоны на основании единого разрешения на продажу.
Эта процедура авторизации позволяет фармацевтическим компаниям подавать в EMA единую заявку на получение разрешения на продажу и продавать лекарство и делать его доступным для пациентов и медицинских работников на всей территории Европейской экономической зоны на основании единого разрешения на продажу.
Заявители могут одновременно подать заявку на получение регистрационного удостоверения ЕС в рамках централизованной процедуры и заключения для использования их лекарства за пределами ЕС. Дополнительную информацию см. в разделе Лекарства для использования за пределами Европейского Союза.
Шаги перед подачей заявки
Представление запроса на участие конкретной форме и сопровождается обоснованием.Когда: с 18 до 7 месяцев до подачи заявки на разрешение на авторизацию маркетинга Дополнительная информация: для подачи заявки |
|---|
Кандидатам следует внимательно отнестись к дате подачи, сверяясь с опубликованными датами подачи и приведенными ниже рекомендациями:
Чтобы уведомить Агентство о предполагаемой дате подачи, заявители должны отправить форму запроса предварительной подачи через службу поддержки EMA, выбрав тип вопроса «запрос на этапе предварительной подачи», а затем «запрос письма о намерениях». Если у вас нет учетной записи EMA, создайте ее на портале управления учетной записью EMA.
Когда: 7 месяцев до подачи заявки на подачу заявки на авторизацию маркетинга Дополнительная информация : Руководство по предварительному авторизации-Раздел 2: шаги до подачи заявки |

Комитет по лекарственным средствам для человека (CHMP) и Комитет по оценке рисков фармаконадзора (PRAC) назначают (со-)докладчиков для проведения научной оценки. В отношении лекарственных средств передовой терапии также назначаются (со)докладчики из числа членов Комитета передовой терапии (CAT), которые будут руководить оценкой. Дополнительная информация: являются лучшей возможностью для заявителей получить процедурные и нормативные консультации от Агентства:
Успешные встречи перед подачей вместе с информацией в руководстве должны позволить кандидатам подавать заявки в соответствии с законодательными и нормативными требованиями. Когда: От 6 до 7 месяцев до подачи заявки на получение торговой лицензии Дополнительная информация: Руководство по предварительной авторизации – раздел 2: Действия перед подачей заявки |
 Это ускоряет процесс проверки.
Это ускоряет процесс проверки.Повторное подтверждение даты сообщений | |||||||
|---|---|---|---|---|---|---|---|
Заявители должны быть повторно пособия, или в соответствии с положениями, или инициированы, инициируемые, или инициированы, инициируемые, инициируемые, или инициируемые, или инициируемые, инициируемые, или не будут представлены, инициированные, или не будут представлены в соответствии с EMASES, или не посвящены. ниже:
Если запланированная дата подачи изменена, кандидаты должны сообщить об этом EMA, повторно отправив заполненную форму запроса на предварительную подачу, указав новую предполагаемую дату подачи в соответствующем поле. Если у вас нет учетной записи EMA, создайте ее через портал управления учетной записью EMA. процесс. Когда: 2–3 месяца до подачи заявки на получение торговой лицензии Дополнительная информация: Руководство по предварительной авторизации – раздел 2: Действия, предшествующие подаче заявки
Assessment of the application
Решение Европейской комиссии по уполномочению маркетинга915159Европейская комиссия по маркетинговой уполномочению151515 Европейская комиссия. | |||||||
Европейская комиссия является уполномоченным органом для всех централизованно авторизованных продуктов, который принимает юридически обязывающие решения на основе рекомендаций EMA. После выдачи Европейской комиссией централизованное регистрационное удостоверение действительно во всех государствах-членах ЕС, а также в странах Европейской экономической зоны (ЕЭЗ) Исландии, Лихтенштейне и Норвегии. Решения Комиссии публикуются в Реестре лекарственных средств для человека. EMA публикует европейский публичный оценочный отчет (EPAR) для каждого лекарства. Когда в новой заявке на получение торговой лицензии отказано, EMA публикует отказ EPAR, включая документ с вопросами и ответами и отчет об оценке. Когда: в течение 67 дней с момента получения мнения CHMP Дополнительная информация: |
 Его необходимо отправить через службу поддержки EMA, выбрав тип вопроса «запрос на этапе предварительной отправки», а затем «уведомление о запросе на изменение».
Его необходимо отправить через службу поддержки EMA, выбрав тип вопроса «запрос на этапе предварительной отправки», а затем «уведомление о запросе на изменение». Цель состоит в том, чтобы убедиться, что все основные нормативные элементы, необходимые для научной оценки, включены в заявку до начала процедуры..
Цель состоит в том, чтобы убедиться, что все основные нормативные элементы, необходимые для научной оценки, включены в заявку до начала процедуры..

Добровольный обмен рынками. Запугивание: Пилотный проект
От 25 марта. для орфанных препаратов и лекарств для лечения рака будут приглашены принять участие в пилотном проекте, заявив о своих намерениях по выходу на рынок на добровольной и конфиденциальной основе.
для орфанных препаратов и лекарств для лечения рака будут приглашены принять участие в пилотном проекте, заявив о своих намерениях по выходу на рынок на добровольной и конфиденциальной основе.
Пилотный проект призван помочь регулирующим органам понять, почему могут возникать задержки в реализации определенных лекарств в государствах-членах ЕС после получения ими разрешения на продажу.
EMA предложит соискателям торговой лицензии поделиться этой информацией с помощью онлайн-опроса во время проверки или при получении мнения CHMP.
Их также попросят предоставить отзывы о проблемах и ограничивающих факторах, с которыми они сталкиваются при обеспечении доступности своих лекарств в ЕС.
Пилотный проект длился 18 месяцев до августа 2022 года. В настоящее время пилотный проект закрыт.
Европейская комиссия, EMA и национальные компетентные органы проводят этот пилотный проект в контексте Фармацевтической стратегии для Европы.